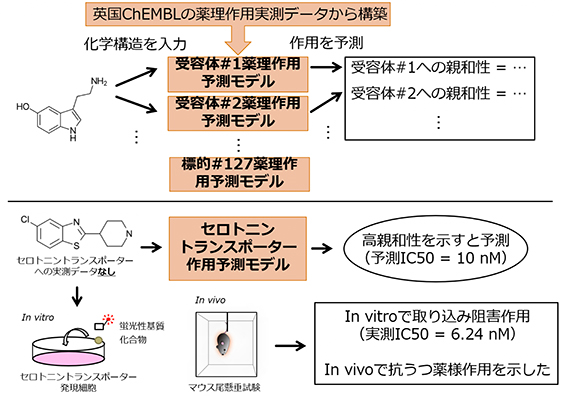
金子周司 薬学研究科教授、永安一樹 同助教、酒井幸 同博士課程学生らの研究グループは、深層学習技術の一手法であるグラフ畳込みニューラルネットワークを用いることで、医薬品の作用標的として特に重要な127種類のタンパク質に対する親和性を化合物の構造情報から予測できる手法を開発しました。さらにこの手法で、抗うつ薬の作用点として知られるセロトニントランスポーターに強く作用する化合物を特定し、その化合物がマウスで抗うつ作用を示すことを見出しました。
これらの結果は、構築した予測モデルの高い妥当性を示していると考えられます。本成果により、目的とする(あるいは目的としない)タンパク質への親和性が計算機上で予測可能となり、目標としていないタンパク質への作用の開発過程における予測・予防や既承認医薬品のドラッグリポジショニングへの応用が期待されます。
本研究成果は、2021年1月12日に、国際学術誌「Scientific Reports」に掲載されました。
【DOI】https://doi.org/10.1038/s41598-020-80113-7
【KURENAIアクセスURL】 http://hdl.handle.net/2433/260969
Miyuki Sakai, Kazuki Nagayasu, Norihiro Shibui, Chihiro Andoh, Kaito Takayama, Hisashi Shirakawa & Shuji Kaneko (2021). Prediction of pharmacological activities from chemical structures with graph convolutional neural networks. Scientific Reports, 11:525.



