2010年10月4日

左から、鈴木特任講師、楠見教授
楠見明弘 物質-細胞統合システム拠点(iCeMS)教授らの研究グループは、細胞内での分子分布の観察のための、細胞分子固定の標準法を新たに確立しました。
研究の概要
生体試料を「固定」してから観察するのは、医学・生物学の領域では250年来の伝統ある方法です。最初は、柔らかい生体試料を「固くして」切断しやすくする目的で、現代的に説明すると、細胞内で「分子が動かないように固定」することが目的で、用いられてきました。18世紀のヨーロッパで始まり、最初はアルコール漬けでした。マリー・アントワネットの主治医であったヴィックダジールも、この方法を最初に記述した一人です。ホルマリン固定は、それから約120年後の1893年にドイツの医師ブルムが始めました。現在でも、医学・生物学分野では、細胞や組織の微細構造や分子分布、および、それらの異常を調べるのは常に重要ですが、最初のステップは、ホルマリン、かつ/または、グルタールアルデヒドなどの分子(化合物)による「化学固定」です。このような現在の細胞分子固定法は、1960年代に、当時大きく進歩しつつあった光学顕微鏡と電子顕微鏡を細胞の研究に応用することを目的として開発されました。
この50年来、この方法が用いられてきましたが、今回の研究で、これでは分子固定が不十分なことが多いことがわかりました。蓄積されたデータすべてについて、再検討が必要になりました。特定の分子をマークして分布を調べるには、その分子の抗体に蛍光分子や微小金粒子などを結合させ可視化しています。ところが1個の抗体は2個の分子に結合できるため、分子固定が不十分で分子が動きうる条件で抗体マーキングをすると、通常では起きない分子の集合が起こっていたのです。さらに、分子の集合は、細胞にとってはある種のシグナルで、その集合部分に、他の未固定分子が集まることも起こります。研究者がこれに気づかないと、「2種類の分子が多数集まって働いている」、という「発見」をしたという誤解が起こります。本研究では、特に、細胞膜上で、シグナル伝達、多くのウィルスの感染や増殖、アルツハイマー病発症などに関わる重要な領域であるラフト領域に関わる分子で、これが顕著なことがわかりました。これは、様々なラフト関連分子の運動を、私たちが開発した最新の方法である「1分子追跡法」で調べた結果わかったものです。固定後は、同じ分子でも様々な固定状態で存在するので、1分子ずつ多数の分子を調べる方法が特に有効でした。
本研究では、さらに、細胞での分子分布を抗体マーキングで調べるための細胞固定法を確立しました。固定操作は強いほど分子運動は止まりますが、抗体が結合しにくくなり、マーキングできなくなります。そこで、両者が両立する条件があることが重要です。固定条件を系統的に変化させ、様々なラフト関連分子がどの程度動くかを調べました。その結果、(1)非常に狭い条件で、両立が可能であること、(2)固定されない分子が、15%程度残ることが多いので、結果の解釈を慎重にする必要、(3)抗体ではなく、抗体から1価のFabという断片を作製して用いるか、GFPなどを用いて生細胞観察をおこなうべきであること、などがわかりました。さらに面白いことに、(4)化学固定だけでなく、固定の原点に戻ったアルコール固定がタンパク質観察に有効であること、もわかりました。
本研究は、楠見明弘 物質-細胞統合システム拠点(iCeMS)教授/再生医科学研究所教授/独立行政法人科学技術振興機構(JST)国際共同研究プロジェクト(ICORP)-膜機構プロジェクト・総括責任者、田中賢治 大学院生(現在、株式会社 味の素)、鈴木健一 JST戦略的創造研究推進事業 さきがけ研究者/iCeMS特任講師らの共同研究として行われました。今回の研究成果は2010年10月3日(米国東部時間)、米科学誌「Nature Methods(ネイチャー・メソッズ)」オンライン版で公開されました。誌面では2010年11月3日号に掲載予定です。
1. 研究の背景と経緯
顕微鏡法における、細胞分子固定の歴史
生体試料を「固定」してから観察するのは、前述のように、医学・生物学の領域では250年来の伝統ある方法です。現在のライフサイエンスでも、細胞や組織の微細構造や分子分布、及び、それらの異常を調べるのは常に非常に重要で、その最初のステップは、ホルマリン、かつ/または、グルタールアルデヒドなどの分子(化合物)による「化学固定」です。化学固定では、「重合性の分子の鎖」を用い、これによって細胞内にあるそこら中の分子、すべてをつなぎ合わせて(架橋をかけて)、結局は、どの分子も動けなくされている(固定されている)と信じられてきました。このような現在の細胞分子固定法は、1960年代に開発されたものですが、実際に、分子が動けないことの確認は、驚いたことに、全くされてこなかったのです。
抗体マーキング(蛍光抗体標識、微小金粒子標識)時におこる、抗体による分子集合の誘起
一方、細胞内での分子分布を調べるには、その分子の抗体を作製し、抗体に蛍光分子や微小金粒子などを結合させることで、分子をマーキングして可視化することによって行います。そのようなデータの一例を、図1に示します。
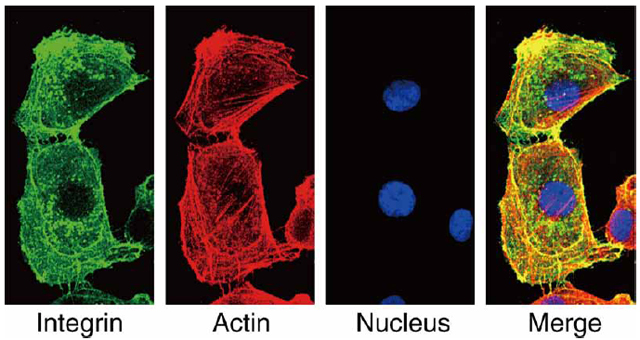
- 図1: 固定後の抗体マーキングの例。ここでは、細胞膜分子のインテグリンが蛍光抗体でマークされている(緑色)。それ以外に、DNAが青色蛍光分子で、細胞骨格のアクチン線維の束が赤色蛍光分子で染色されている。
もし、予想通りには、分子が固定されていないときに、抗体を加えるとどうなるでしょうか?1個の抗体は2個の分子に結合できるため、分子が動きうる条件で抗体マーキングをすると、2個の分子が集まります。通常の抗体を用いると、1つの対象分子の違う2カ所、3カ所・・・に結合する抗体を含んでいるので、分子の集合はどんどん進みます。また、多くの可視化法では、抗体を結合させるだけではシグナルが弱すぎるため、抗体を結合させたあと、その抗体に対する抗体を標識しておき、それをさらに加えて、2段階でシグナルを増幅するという方法が一般的です。そうすると、分子が動ける状態で抗体マーキングをすると、大きな分子集合体が生じてしまうのです(図2)。
この問題は、膜分子で顕在化することが予想できました。細胞膜は液体なので、その中で分子は動いているし、2次元という低次元性のために、分子同士の衝突頻度が非常に高いことが予想されるからです。そういう条件で、抗体をかけたり、さらに2次抗体(2番目にかける抗体に対する抗体、蛍光や微小金粒子で標識してある)をかけたりすると、非常に大きな、会合体ができるのです(図2)。
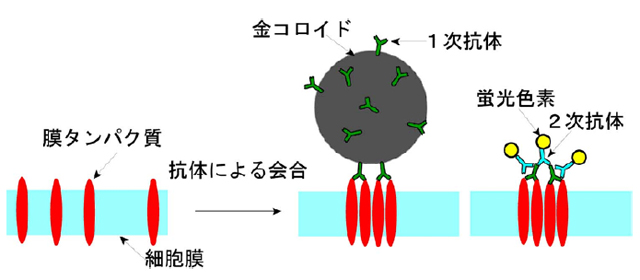
- 図2: 抗体が、細胞膜分子の集合を誘導する様子。
細胞質の中で浮いている分子を調べるのには、抗体マーキングとか固定は、あまり使われてきませんでした。3次元の水中で動き回っていて固定効率が悪いことが予想され、また、細胞内の特定の場所へ濃縮することなども少ないと仮定されていて、その結果、これらの分子については、研究自体がほとんどありません。
また、多くの分子は、核中で核酸に結合したり、細胞質中で細胞骨格に結合したりしています。また、細胞骨格や染色体は、多くの生体高分子が集合してできています。これらが、もとの構造を保ったまま固定できているかどうかは、大きな問題ですが、これらには多数のタンパク質が会合しているため、「重合性の分子の鎖」はそれなりに有効であると考えられます。
それで、最も大きな問題は、細胞の膜分子の分布にあるということになります。
細胞中にあるタンパク質分子の約40%は膜にあり、細胞の反応の半分以上が膜で起こります。
細胞膜中のラフト領域研究で問題が大きく顕在化してきた
細胞膜は2次元の液体ですが、理想的に全ての分子が混じり合った液体ではありません。溶媒同士ですでに分子間の相互の混じり合い方にバリエーションがあり、一様になれずに色々なかたまりが生じていると考えられています。このなかで、もっとも一般的で有名なものは、ラフト領域と呼ばれるものですが、名前だけが一人歩きしている状況で実体がほとんどわかっていません。ある程度わかっているのは、細胞外からラフト領域関連の受容体を刺激した場合で、そのときには、「受容体は会合し」、会合した受容体にはラフト関連脂質が集まってきて、安定化された(寿命が1分以上)10nm以上の大きさの「安定化ラフト」が形成されます。これは、顕微鏡で見ることが可能です(図3)。
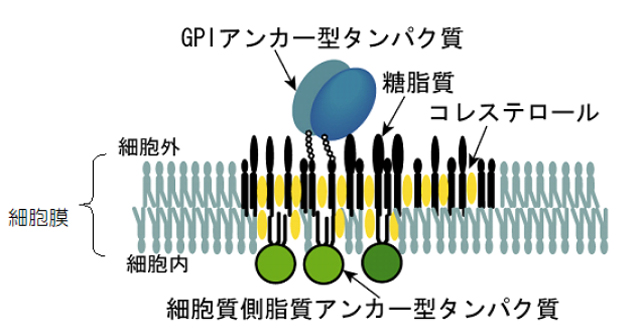
- 図3: 細胞外からGPIアンカー型受容体を刺激すると、安定化ラフトが生じ、そこに、細胞内のラフト関連シグナル分子が集まってきた様子を示す模式図。細胞外からGPIアンカー型受容体を刺激すると、受容体は会合し、そこにはラフト関連脂質が集まってくる(コレステロール、糖脂質など)。このようなラフトはGPIアンカー型受容体の会合によって安定化されている(寿命が1分以上)。これは、顕微鏡で見ることが可能である。そこに、細胞内の脂質アンカー型タンパク質がリクルートされてくる。
- 最近までは、抗体による人工的会合を(それと気づかず)見ていたため、刺激以前にも、直径百~数百nm 程度の大きさの安定な構造を持つラフトと呼ばれる領域が多数存在していて、その構造にいろいろな分子が取り込まれてシグナル伝達が起こるという考えが大勢を占めていた。本研究結果により、これは、誤りであると明らかになった。
しかし、刺激前にラフトがあるのか、あるとしたらどのようなサイズと寿命か、どのような分子からできているのかなど、全くわかっていませんでした。それで、世界中の研究者が、顕微鏡で刺激前のラフトを見よう、ラフトにある分子を蛍光抗体法や金粒子標識でどこにあるか調べようと、1990年頃から躍起になり始めました。しかし20年近く経っても、共通の認識が得られていません。実験の多くは、蛍光抗体で染色して分子の分布を調べるという、古典的な、今では学部4年生の実験でもできるような、簡単なものなのです。にもかかわらず、刺激前の細胞のラフトはどのようなものか(存在するのか)について、再現性がとれない報告が山と積まれる研究領域になってしまったのです。
私たちは、それらの論文を調べたり、自分たちで実験している間に、以下の仮説に到達しました。
- 普通の条件でおこなわれた化学固定ではラフト関連分子は固定されにくい。特に、「脂質アンカー」と呼ばれる、脂質によって膜に結合しているタンパク質は固定されにくい。
- 固定が不足しているので、抗体によってターゲットの分子Aに会合が誘導される。
- 細胞膜(2次元の液体)の溶媒である脂質は、ほとんど固定できない(拡散は遅くなる)。
- ターゲットの分子Aがラフト分子の時、それが抗体によって会合させられると、そこにラフト関連の脂質分子(固定されない)が集積し、上で述べたように安定ラフトが形成されてしまう。その安定ラフトに、他のラフト関連シグナルタンパク質B, C, D・・・が集まる。すなわち、図3と同じような状況が人工的に作り出され、これらのタンパク質は、シグナルが来る前から複合体を形成しているという、誤った「大発見」につながる。
- 同じような固定条件で実験しても、化学固定試薬の有効濃度、反応温度、細かい抗体反応条件によって、分子の集まり方が異なる。実際、同じ培養皿中の多数の細胞を調べると、その中でも、分子の集まり方が大きくばらついていることが多い。このようなバラツキは、正確に報告されていない可能性がある。
私たちは、総説にもこのようなことを書いて、「だからいがみ合うな。もっと厳密に実験を」と言ったのですが(例えば、Kusumi A. and Suzuki K., Biochim. Biophys. Acta, 1746, 234-251, 2005には、このことが一番詳しく記述されている)、固定の程度についての定量的結果が文献にないこと、どのような固定がよいかという細胞分子固定法が確立されてなかったので、実効については疑わしかったのです(国際シンポジウムなどでは、この議論は、討論中などに引用され、研究者の関心は高まっていました)。
そこで、このコンテクストで、ラフト分子を中心とした実験をすることにしたのです。
2. 研究の内容
研究のクリティカルポイント
以下の3点が、本研究の、クリティカルなポイントです。
- 固定条件を強くしたら、細胞分子固定は可能か?
- 固定条件を強くすると、抗体は分子を認識できなくなる。パラホルムアルデヒドだと5%(室温、1時間程度)、グルタールアルデヒドだと0.2%(室温、30分程度)程度が限度(この範囲でも、固定条件を強くすると、抗体の結合は、どんどん減少していく)。固定と抗体による認識を両立できる固定条件が見つかるか?
- 固定操作をすると、分子の動ける程度には、大きなバリエーションが生じる。平均値には意味がない(例えば、70%の分子は運動停止しているが、残りの30%の分子は元の半分の拡散係数で運動している、という具合に調べる必要がある)。それで、1分子毎に追跡する。これを多数の分子について繰り返す。
調べた分子
調べた分子を図4に示します。すべて、細胞膜中の分子です。左端のトランスフェリン受容体と右端の(膜の溶媒を作る基本分子である)リン脂質はラフトに入らない分子、それ以外はすべて、ラフト関連分子と考えられています。
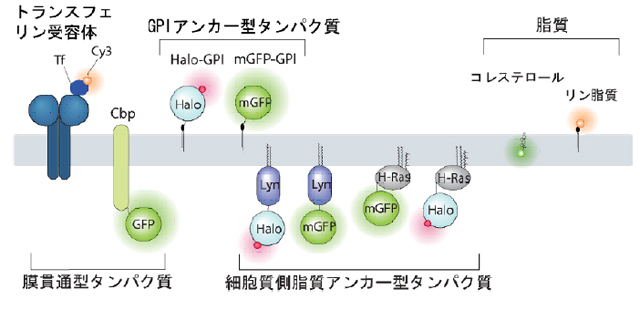
- 図4: 本研究で検討した膜分子。左端のトランスフェリン受容体と右端のリン脂質は、非ラフト分子、それら以外は、ラフト関連分子である。両者の間に著しい違いはなかった。
研究のクリティカルポイント(最初の2点)に対する回答(図5, 6)
- 固定条件を強くしたら、細胞分子固定は可能か?
- 膜貫通型タンパク質分子は、固定を強くすると、固定は可能。
- GPIアンカー型タンパク質といわれる主要なラフト関連タンパク質は、普通の化学固定(抗体の結合能をある程度保てる条件の固定)では、固定は不可能。
- GPIアンカー型タンパク質は、固定の原点に戻って、アルコール固定すると、固定可能。しかし、18世紀とは違って冷凍庫があるので、-40℃に冷やしたメチルアルコールを用い、固定中の細胞温度を -20℃くらいにして固定するのがマイルドでよい(5分以内、抗体の結合能が保たれやすい)。
- 脂質はラフト関連分子も非ラフト分子も化学固定は不可能。アルコール固定をすると、ほとんどの脂質が抽出されて、細胞から無くなるので、実用的でない。
- 細胞分子が固定され、かつ、抗体が認識して結合するという条件は見つかるか?
- 非常に狭い条件で見つかった。化学固定だと、4%パラフォルムアルデヒド + 0.2%グルタールアルデヒド(許容範囲は、3.6~4.4%パラフォルム、0.1~0.2%グルタール)、25℃前後、30分(60分では苦しい)。
- アルコール固定は、上記の通り。しかし、細胞からの背景蛍光が増えるので、化学固定がダメなときに限る。
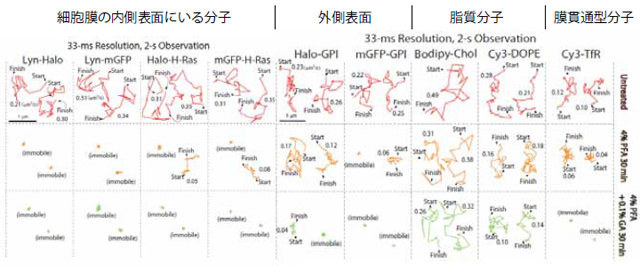
- 図5: 様々な膜分子について、2種の細胞分子固定操作をおこなったあとの拡散運動。典型的な軌跡を示す。
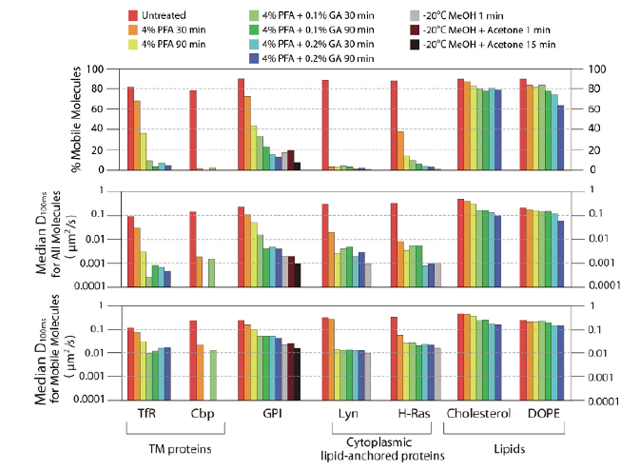
- 図6: 様々な膜分子の、色々な細胞分子固定をおこなったあとの、拡散運動。可動成分(上)、拡散係数の中央値(中)、可動成分の拡散運動の中央値(下)を示す。
その他の重要ポイント
- 上記の方法を用いても、固定されない分子が5~20%程度残ることがわかりました。動ける分子は、マーキング用の抗体によって集められることを念頭に置いた結果の解釈が必要なことがわかりました。
- 従って、上記の固定条件を用いても、抗体マーキングには、抗体から1価のFabという断片を作製して用いるか、GFPなどを用いて生細胞観察をおこなうべきであることがわかりました。
- 固定が上記より弱いと、抗体マーキングによって、分子の会合が起こることを直接に証明しました。これは、この50年来蓄積されたデータすべてについて、再検討が必要なことを示しています。
3.今後の展開
新しい細胞分子固定法が確立されたことで、細胞の膜研究の進展がスムーズになることが期待されます。一方、ここでの結果を参考にして、これまでに蓄積された抗体マーキング法のデータのうち、信頼できるものと、疑わしいものを区別することが可能です(新しいものに、危うい条件のものが多いことは、面白い特徴です)。
特に、細胞膜上のナノ・メゾスケールドメインであるラフト領域の構造・機能の解明、さらには、ラフト経由の感染・増殖や発病過程の解明につながることが期待されます。
例えば、ラフト領域は、BSE(牛海綿状脳症)やインフルエンザやエイズウィルスなどのウィルスの感染や増殖、アルツハイマー病の発症などにも関わっていると考えられていますが、これらの研究の大きな障害の一つ(抗体マーキングが機能しないという障害)が取り除かれたことになります。今後の、この分野の研究の進展が期待されます。
用語解説
1)固定法を作った人たち
 | Félix Vicq-d'Azyr(ヴィックダジール) 固定法は、組織学、解剖学、薬理学の歴史を作ってきた方法でもある。最初は、柔らかい生体試料を「固くして」切断しやすくする目的で、現代的に説明すると、細胞内で「分子が動かないように固定」することが目的で、用いられてきた。最初におこなわれたのは、アルコール漬けにする方法で、18世紀の後半にヨーロッパで始まった。マリー・アントワネットの主治医であったヴィックダジールも、この方法を最初に記述した一人である。 |
Blum F
ホルマリン固定は、それから約120年後の1893年にドイツの医師ブルムが始めた。
Karnovsky MJ
現在の化学固定法は、1960年代に、カルノフスキーを代表とする多くの人々によって確立された。この方法では、ホルマリン、かつ/または、グルタールアルデヒドなどの分子(化合物)の溶液を用いる。これらの分子は、重合性の分子で、細胞内の分子と共有結合して、ほとんど全ての分子をつないでしまおうとするものである。これは「化学固定」と呼ばれる。当時大きく進歩しつつあった光学顕微鏡と電子顕微鏡を細胞の研究に応用することを目的として開発された。
2)ラフト
細胞膜は2次元状の液体である。それで、細胞膜を海にたとえると、そこにぷかぷか浮いているイカダ、というイメージからラフトという名前が使われるようになった。細胞膜は一様ではなく、氷山のようなものができていたり、シャーベット状の塊(正しくは、秩序液晶相という領域)があったりと考えられている。しかし、2次元の液体という中での、協同的現象として現れるドメイン構造は、はっきりと捉えることが難しく、この議論は40年間続いている。
人工膜では、このような膜領域を作り出すことができている。しかし、実際の細胞膜がどうなっているかは、ほとんどわかっていない。人工膜では、大抵、2-3種類の脂質分子で膜を構成するので話は簡単であり、そこで、シャーベット状の塊を作るために必要な代表的な脂質分子は、コレステロールと糖脂質であることもわかっている。しかし、細胞膜中には1万種程度の分子が存在し、それらがどのように混ざり合って、液体中に不均一な構造を作るのかは、ほとんどわかっていない。
この難しさに混乱を加えたのが、免疫蛍光法や免疫電顕法と呼ばれる抗体マーキングによる観察結果である。今回の研究で、この混乱は、化学固定が不十分な分子が、抗体によって集められることが原因であることがわかった。
3)生細胞中での1分子追跡
生細胞中の目的分子に、蛍光分子や直径40nm程度の金微粒子の目印を結合させ、それらを、様々な光学顕微鏡を用いて1分子毎に追跡する。In vitroでの(ガラス上に取り出した生体高分子などを用いておこなう)実験に比べて、以下の点が難しい。(1)細胞には蛍光を発する分子が多数存在し、また、微粒子と紛らわしい顆粒が多数存在する。蛍光を減らす工夫が必要、(2)細胞に対する光毒性への注意が必要、(3)蛍光の褪色を防ぐための分子状酸素の除去は、普通はやらない方がよい、(4)細胞を活かしておくための道具立てが必要で面倒、など。細胞をよい状態に保ちながら、刺激などをおこなって面白い瞬間を捉えるためには、観察の自動化、画像解析の自動化、ノイズの中からシグナルを拾って1分子を追跡するためのソフトウェアの開発なども重要である。私たちはこの技術開発を1989年以来やってきており、時間分解能は、金微粒子による1分子追跡では4マイクロ秒、蛍光1分子追跡では100マイクロ秒と、他をはるかに引き離している。
掲載論文名
Membrane molecules mobile even after chemical fixation
化学固定後でさえも、膜分子は動くことができる(参考訳)
関連する科学技術振興機構のプログラム
科学技術振興機構 戦略的創造研究推進事業 さきがけ研究
領域名: 生命システムの動作原理と基盤技術 (領域総括:中西重忠)
研究代表者: 鈴木健一
研究実施期間:平成20年10月~平成24年3月
また、本研究は、科学技術振興機構 戦略的創造研究推進事業 国際共同研究プロジェクト 「膜機構プロジェクト」(平成17年3月~22年3月)の一環としておこなわれました。インド側代表者の、Satyajit Mayor教授も、共著者の一人です。
iCeMSウェブサイトでのニュースリリース
http://www.icems.kyoto-u.ac.jp/j/pr/2010/10/04-nr.html
関連リンク
- 論文は、以下に掲載されております。
http://dx.doi.org/10.1038/nmeth.f.314
- 京都新聞(10月4日 22面)および日刊工業新聞(10月4日 18面)に掲載されました。