平成20年6月12日
国立大学法人 京都大学
大学共同利用機関法人 情報・システム研究機構 国立遺伝学研究所
大学共同利用機関法人 情報・システム研究機構 国立情報学研究所
独立行政法人 理化学研究所
国立大学法人 京都大学、大学共同利用機関法人 情報・システム研究機構 国立遺伝学研究所、大学共同利用機関法人 情報・システム研究機構 国立情報学研究所、独立行政法人 理化学研究所は、米国JGI(連合ゲノム研究所)、スクリプス研究所、オックスフォード大学など5カ国、18研究機関との共同研究でナメクジウオ(Branchiostoma floridae)のゲノム解読に成功しました。この国際プロジェクトは、佐藤矩行京都大学教授をリーダーの一人として企画され、国内では文部科学省科学研究費特定領域研究「ゲノム4領域」の支援を得て実施されたものです。この成果は、6月19日発行の英国科学誌Nature(ネイチャー)誌に掲載されました。
ナメクジウオは同じ脊索動物であるヒトなどの脊椎動物の祖先と考えられ、古くから研究の対照となってきた生物です。今回のナメクジウオ全ゲノム解読により、約5億塩基対の全ゲノム配列の中に、ヒトの遺伝子組成とよく似たおよそ21,600個の遺伝子が見つかりました。
今回のナメクジウオゲノムの解読により、脊椎動物も含まれる脊索動物の共通の祖先の特徴を明確にすることができました。このことは、ダーウィンの進化論発表以来長い間論議されてきた、脊椎動物の起源の解明に大きな一石を投じた成果といえます。
 |  |
| (図:今回のゲノム解読の対象となった、フロリダナメクジウオ (川島武士博士提供)) | |
ナメクジウオは脊索動物の一種、頭索動物に分類される生物で、熱帯と温帯に広くに生息し、約30種類が記載されています。日本近海にはこれまでに3種類が確認されていますが、様々な地域集団が存在すると予想されています(参考文献:安井、窪川)。
ナメクジウオはヒトなど脊椎動物の祖先と考えられ(脊椎動物は脊索動物に含まれます)、古くから研究の対象となっていました。脊椎動物がどのように進化してきたのかという問題について、これまでは、脊索動物の中で一番最初に現れたのがホヤの仲間(尾索類)で、次にナメクジウオの仲間(頭索類)、そして脊椎動物が現れたと考えられていました。また、脊椎動物の進化に伴って全ゲノムの重複が2回おこり、それが複雑な体づくりを可能にしたのではないかという大野らの提案もありました。
今回のナメクジウオゲノムの解読は、脊椎動物の進化についてダーウィンの進化論発表以来の懸案となっていた問題を一気に解決し、脊索動物の進化と脊椎動物の起源を明らかにするものです。この成果をもとに脊索動物、脊椎動物の進化についての研究が飛躍的に発展することが期待されます。
研究成果の概要
(1) 本研究では、ナメクジウオ全ゲノム約5億塩基対の配列を決定し、その中におよそ21,600個の遺伝子を見つけました。ナメクジウオゲノムはヒトゲノムの約6分の1の大きさですが、ヒトの遺伝子組成ととてもよく似ていることが明らかになりました。今回のゲノム解読には単一個体の精子から抽出したDNAを用いたのですが、同じ個体に含まれる二組のゲノムの間に3.7%の塩基の違い(1塩基多型、SNP)が見つかり、これに挿入・欠失の違い6.8%を加えると、多型を示す領域がゲノム全体の10.5%を占めることがわかりました。この割合は、これまでに調べられた生物種の中では最高の値です。同一地域で繁殖し、種としての集団を保っている生物のゲノムが、このように大きな多型性を示すことが初めて明らかにされたことにより、生物の進化と種の確立、集団としての繁殖と種の維持といった、生命現象の根幹に関わる新しい問題を提起するものです。
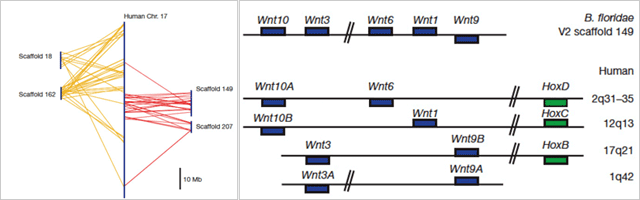
(2) 次にナメクジウオゲノムは脊椎動物への進化の痕跡を非常によく残しており、染色体上における遺伝子の並び順(シンテニー)がナメクジウオと脊椎動物のゲノム間で非常によく保存されていることが明らかになりました。これは、脊椎動物の起源を考える上で非常に重要な情報です。ナメクジウオの染色体上における遺伝子の並びをより詳細に解析することにより、脊索動物染色体の基本型ともいうべき17本の染色体構成を明らかにすることにも成功しています。既にゲノム解読が報告されている尾索動物のホヤでは大規模なゲノムの再構成と脱落がおきているため進化的な解析が困難でした。
(3) シンテニーの解析から、ナメクジウオゲノムの1箇所の染色体断片が脊椎動物ゲノム内での4箇所の染色体断片に対応することが多いことがわかり、脊椎動物の進化に伴ってゲノムレベルでの重複が2回おこったことが初めて確実に証明されました。
(図左:ナメクジウオゲノムとヒトゲノムとのシンテニー、図右:4倍加したゲノム領域の例)
(4) また、今回のゲノム解読から明らかになった遺伝子のうち1090個について比較解析研究を行うことにより、脊索動物の中でナメクジウオが最も初期に進化したことがわかりました。ゲノム全体を対象とした網羅的な解析が行われたことにより、ほぼ完全な証明が得られたものです。このため、より原始的な脊索をもたない動物との進化関係を非常にうまく説明することができるようになりました。
本研究による全ゲノム比較により、尾索動物、頭索動物と脊椎動物という3つの脊索動物グループ間について曖昧だった関係を明確にし、またこれら脊索動物の共通の祖先の特徴を明らかにできたのです。まさに脊椎動物の起源に大きな一石を投じた研究成果といえるでしょう。
 | → |  |
(5) 本プロジェクトを遂行してゆくにあたり、多型性の大きなゲノムについて全配列の解読を可能にするバイオインフォマティクスの新しい手法も開発されています。ナメクジウオゲノム情報は、米国ではJGI(http://genome.jgi-psf.org/Brafl1/Brafl1.home.html)からオンライン提供されており、国内では国立遺伝学研究所で運用されている日本DNAデータバンク(DDBJ:http://www.ddbj.nig.ac.jp)から公開される予定です。
研究の背景
ナメクジウオは脊索動物門頭索動物亜門に属します。同じ脊索動物門には、ホヤ等が含まれる尾索動物亜門、また、進化的により高等な脊椎動物が含まれます。特徴を箇条書きにしてみましょう。
- (1) 体長は30-50mm程度(時に70mmに達する)で、外見は魚類に似ています。
- (2) 頭部を持たず、前方腹側には摂食器を覆う口被蓋という器官が発達しています。
- (3) 体の前半部にある鰓列という構造で水中の酸素を取り込み、食物を濾しとることで栄養を得ています。
- (4) 閉鎖血管系ですが心臓はありません。
- (5) 光受容器官はありますが目に相当する器官はありません。
- (6) 背びれ、腹びれに相当する器官は目立ちませんが、尾びれに相当する器官が発達しており、よく泳ぎます。
- (7) 頭部から尾部まで筋肉組織でできた棒状の組織「脊索(notochord)」を持ち、このため頭索動物とよばれています。脊椎動物では発生の過程で一度つくられた脊索が退化して脊椎に置き換わりますが、ナメクジウオでは、脊索のまま残ります。
- (8) 脊索に付随した神経索(神経管、nerve cord)を持ちますが、神経節と脳はありません。雌雄があり、繁殖期には生殖腺をみることができます。メスの生殖腺は黄色っぽいのに対し、オスの生殖腺は白いことで判別できます。
(以上の参考文献:安井、窪川)
実験動物としてのナメクジウオの歴史は古く、ドイツのP. S. Pallasが1774年に記載したのが最初とされます。しかしその後、環境の変化などで生息地が狭められ入手が困難となったことなどから、研究対象としてあまり注目されなくなりました。1990年代に入り、分子生物学を駆使した比較発生学の分野で再度実験材料として見直され、多数の遺伝子の単離とその発現解析が進められましたが、入手の困難さと実験における扱いにくさから、そうした研究が飛躍的に進んでいるとはいえません(参考文献:安井、窪川)。しかし、脊椎動物の祖先と進化を知る上では非常に重要な生物であり、実験動物として、より一般化するためにはそのゲノムの解読は不可欠です。このことから、研究者コミュニティの意見も受け、文部科学省科学研究費特定領域研究―ゲノム4領域-の支援を得て2005年(平成17年)からナメクジウオゲノムプロジェクトを開始しました。
このプロジェクトは文部科学省科学研究費補助金特定領域研究の計画研究課題の一つとして行われています。特定領域研究「ゲノム」(2005-2009年度)では生物学、医学、コンピュータ科学、理工学、など広い範囲から研究者が集まり、積極的に融合的な共同研究を推進しており、バイオインフォマティックスの独自手法の開発や、ゲノム配列解析などを集中的に引き受ける研究支援グループをつくり研究の高度化と効率化を図っています。
ナメクジウオゲノム配列自体は2007年には完成し、研究コミュニティへオンラインで公開してきました(http://genome.jgi-psf.org/Brafl1/Brafl1.home.html)ので、世界中で盛んに活用されています。この配列をさまざまな動物のゲノムと比較し、その成果を合わせて今回の論文発表に至った次第です。
研究内容の詳細
1) ゲノム解読と遺伝子予測
ナメクジウオのゲノム解読は全ゲノムショットガン法で行いました。解読したゲノム配列の総塩基対数は約5.2億で、染色体の構成や連鎖地図が未知のため、19対の染色体上への正確なマッピングは出来ませんでしたが、総塩基対数の11.5倍に及ぶ配列情報を解読し継ぎ合わせることで、約95%について正確に解読でき、進化を論じるのに十分な長い断片へと再構成することができました。さらに遺伝子の先頭の配列を効率的に収集する手法を利用して、その後ろにある遺伝子本体の構造を予測するコンピュータプログラムを用いて約21,600個の遺伝子を予測しました。ここから脊索動物で共通の8,437個の遺伝子ファミリーが見いだされ、ナメクジウオの13,610個の遺伝子、ヒトでは13,401個がこのファミリーに属する保存された遺伝子であることが明らかになりました。また、脊索動物には共通でそれ以前の動物には見られない遺伝子が239個見いだされました。
2) 脊椎動物ゲノム進化の推定
さらに、上記の染色体断片を他の動物のゲノムと比較することで、脊椎動物とナメクジウオとでどの部分がどれだけ類似しているかも広範囲に分析することも可能になり、脊椎動物ゲノムがどのように進化してきたか、その道筋が見えてきました。
1970年、大野乾(すすむ)は著書「Evolution by Gene Duplication(遺伝子重複による進化)」の中で、脊椎動物ではゲノム全体が重複する現象が複数回起き、進化に有利に働いたという全ゲノム重複説を提唱しました。多くの生物学者に多大な影響を与えた説ですが、その信憑性は論争の的でした。幸い21世紀に入り脊椎動物ゲノムの解読は進展し、大野の全ゲノム重複説は検証可能になってきました。2007年に日本のゲノム研究グループが報告したメダカゲノム解読によって硬骨魚類出現以降のゲノム進化の様子は明らかにされていましたが、2002年に報告されたホヤのゲノム解読では、全ゲノム重複が起こった可能性を示唆する結果は得られたものの、これが全ゲノム重複によるものか、断片的な重複で起きたのかを十分に断定できませんでした。
今回、ナメクジウオゲノムの解読に基づき、計算機を用いてヒト、ホヤ、ナメクジウオのゲノムを比較解析した結果、ヒトとナメクジウオで保存された17の染色体断片(連鎖群)を見出すことができ、ヒト遺伝子の約90%がこの連鎖群に含まれていました。脊索動物はこの17本の染色体を基本とするものと推定されます。また、ヒトゲノム上にこれらの連鎖群がそれぞれ4ヶ所で見出され、脊索動物から脊椎動物が進化するにあたり2回の全ゲノム重複が起きたとする大野の仮説が全ゲノムの情報をもとに裏付けられたのです。この全ゲノム重複は尾索類の分岐ののち、軟骨魚と硬骨魚が分岐する以前に起きたと推測されました。この時代から現代まで生き残っている脊索動物は他に無顎類だけで、そのうちのヤツメウナギについては、2回の全ゲノム重複の間で分岐した可能性が示唆されています。
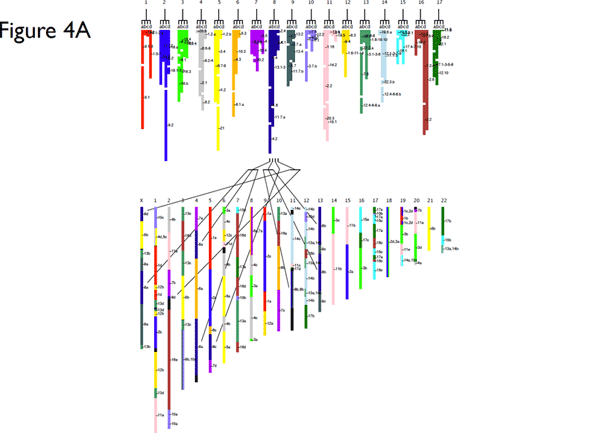
(図:脊索動物染色体構成の原形とヒト染色体の関連)
また、脊索動物共通の祖先から、頭索動物のナメクジウオが最初に分岐し、その後、尾索動物のホヤが分岐したこと、ホヤでは大規模なゲノムの再構成、脱落が起きてゲノムが小型化していたため、全ゲノム重複が分かりにくくなっていたことが確認できました。さらに、ナメクジウオのゲノムが驚くほど太古の脊索動物の共通祖先のありようを今に残していることを示唆する結果となっています。また、ナメクジウオとヒトで、遺伝子を制御する非翻訳配列が緩やかに保存されていました。これらの配列はこれまでに直接見出された最古の非翻訳領域といえます。これは脊索動物の祖先の遺伝子制御のあり方を理解する上でとても興味深いデータです。
3) ナメクジウオゲノムの多様性
ナメクジウオには様々な地域集団が存在します。今回は主としてフロリダ由来系統のゲノム解読を行ったのですが、同じ個体由来のゲノムでありながら非常に多型に富むことが明らかとなりました。3.7%の塩基の違い(1塩基多型、SNP)と、挿入・欠失多型6.8%を加え、多型を示す領域がゲノム全体の10.5%を占めています。この値は、これまでに報告された生物種の中では最高の値となっています。
4) 論文タイトル、著者等
The amphioxus genome and the evolution of chordate karyotype
Nicholas H. Putnam1,2, Thomas Butts3, David E. K. Ferrier4, Rebecca F. Furlong3, Uffe Hellsten1, Takeshi Kawashima2#, Marc Robinson-Rechavi5,6, Eiichi Shoguchi7#, Astrid Terry1, Jr-Kai Yu8, E`lia Benito-Gutie´rrez9, Inna Dubchak1, Jordi Garcia-Ferna`ndez10, Jeremy J. Gibson-Brown11#, Igor V. Grigoriev1, Amy C. Horton11, Pieter J. de Jong12, Jerzy Jurka13, Vladimir Kapitonov13, Yuji Kohara14, Yoko Kuroki15, Erika Lindquist1, Susan Lucas1, Kazutoyo Osoegawa12, Len A. Pennacchio1, Asaf A. Salamov1, Yutaka Satou7, Tatjana Sauka-Spengler8, Jeremy Schmutz16, Tadasu Shin-I14, Atsushi Toyoda15, Marianne Bronner-Fraser8, Asao Fujiyama15,17, Linda Z. Holland18, Peter W. H. Holland3, *Nori Satoh7# & *Daniel S. Rokhsar1,2
* Corresponding authors
- 1 Department of Energy Joint Genome Institute, Walnut Creek California 94598, USA.
- 2 Center for Integrative Genomics, Department of Molecular and Cell Biology, University of California, Berkeley, California 94720, USA.
- 3 Department of Zoology, University of Oxford, South Parks Road, Oxford OX1 3PS, UK.
- 4 The Gatty Marine Laboratory, University of St Andrews, St Andrews, Fife KY16 8LB, UK.
- 5 Department of Ecology and Evolution, University of Lausanne, 1015 Lausanne, Switzerland.
- 6 Swiss Institute of Bioinformatics, 1015 Lausanne, Switzerland.
- 7 Department of Zoology, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan.
- 8 Division of Biology, California Institute of Technology, Pasadena, California 91125, USA.
- 9 National Institute for Medical Research, Mill Hill, London NW7 1AA, UK.
- 10 Departament de Gene`tica, Facultat de Biologia, Universitat de Barcelona, Avenue Diagonal, 645, Barcelona 08028, Spain.
- 11 Department of Biology, Washington University, St Louis, Missouri 63130, USA. 12Children’s Hospital of Oakland Research Institute, Oakland, California 94609, USA.
- 13 Genetic Information Research Institute, 1925 Landings Drive, Mountain View, California 94043, USA.
- 14 National Institute of Genetics, Mishima, Shizuoka 411-8540, Japan.
- 15 RIKEN Genomic Sciences Center, 1-7-22 Suehiro-cho, Tsurumi-ku, Yokohama, Kanagawa 230-0045, Japan.
- 16 JGI Stanford Human Genome Center, 975 California Avenue, Palo Alto, California 94304, USA.
- 17 National Institute of Informatics, 2-1-2 Hitotsubashi, Chiyoda-ku, Tokyo 101-8430, Japan.
- 18 Marine Biology Research Division, Scripps Institution of Oceanography, University of California San Diego, La Jolla, California 92093-0202, USA.
- # Present addresses: Okinawa Institute of Science and Technology
(OIST), Uruma, Okinawa 904-2234, Japan (T.K., E.S. and N.S.); 909 Hiawatha Drive, Mount Pleasant, Michigan 48858, USA (J.J.G-B.).
今後の展開
我々ヒトは脊索動物に属し、そこには脊椎動物の他に頭索動物、尾索動物が含まれます。姿、かたちは大きく違っていますが、種を越えた共通のメカニズムを使っていることが最近の研究からわかってきました。また、互いに比較することでそれぞれがどのように進化してきたのか、共通の祖先はどんな生物だったのか、といった問題についても研究が進められており、これから数年の間に大きな進展が期待されています。今回明らかになった2回の全ゲノム重複のあと、脊椎動物にどのようなゲノム進化が起こったかは、興味深い研究テーマです。
化石の研究からは、形態などの特徴の変化を伴う種分化と、全ゲノム重複との関係を明確に示すことは困難でした。しかし、2回の全ゲノム重複によって脊椎動物の祖先のゲノムが作られたことが、脊椎動物の特徴(軟骨形成や骨格形成の神経堤細胞、体節(硬節による区画)、精巧な後脳のパターン、精密な神経系、複雑な内分泌系)をもたらした可能性はあります。事実、4倍に増えた遺伝子のうち、発生におけるシグナル伝達や遺伝子制御を行う遺伝子が、他の遺伝子に比べて圧倒的に多数のまま残されてさまざまな働きをしていることは、その可能性を支持するものでしょう。
一方、ゲノムを解読する装置についても最近の3、4年で大きな技術革新がありました。単位時間あたりの塩基解読量がそれ以前の技術と比べ2~3桁改善されるという画期的な進歩が続いており、今後のゲノム解読スピードは飛躍的に上昇すると考えられます。1塩基多型(SNP)を同定し収集する速度も大幅に向上し、さらには遺伝子の転写量に影響をあたえるゲノム構造の変化を詳細に分析することも可能になりつつあります。今後、ナメクジウオゲノム解読を通じて培ったゲノム解析用ソフトウエア技術が活かされることでしょう。
用語解説
大野 乾(1928-2000)
1953年北海道大学で博士号取得後渡米。City of Hope National Medical Center の研究者。染色体進化、性染色体の研究分野で顕著な業績があり、特に1970年に出版された著書「Evolution by Gene Duplication(遺伝子重複による進化)」は広く読まれています。
ゲノム
ある生物が持つすべての遺伝情報の一セットを指します。デオキシリボ核酸(DNA)という物質で構成され、4種類のヌクレオチド(A、T、G、C)の重合体です。この4種類の塩基の並び(配列)によって遺伝情報が決定されます。ナメクジウオは約5億塩基からなるゲノムを持っていました。
シンテニー
2つの生物のゲノムの間で、染色体上のある程度の数の遺伝子の並びが類似している状態をいいます。シンテニーは既知のゲノム上での遺伝子の位置情報から、別のゲノムの遺伝子位置を推定するときなどに使われます。シンテニーは生物のなりたちを知る上で有効かつ重要な情報となっています(参考文献:藤山)。
脊索動物・脊椎動物・頭索動物・尾索動物
動物は一般的に、背骨をもつ脊椎動物と、背骨を持たない無脊椎動物に分けられます。しかし、脊椎骨の発生過程を詳しくみると、脊索という基本構造からできてきます。しかも脊索はホヤなどの尾索動物(幼生の尾に脊索がある動物)やナメクジウオなどの頭索動物(脊索が頭の先端まで伸びている動物)にもあることから、現在の動物分類学では、これら3群をまとめて脊索動物とし、これを一つの門としています。すなわち、我々は脊索動物門・脊椎動物亜門、ホヤなどは尾索動物門・尾索動物亜門、ナメクジウオなどは脊索動物門・頭索動物亜門です。最近の化石記録から、これらの動物は今から5億2千万年以上昔に、共通の祖先から進化してきたものと考えられています。
全ゲノム重複
ゲノム全体が重複される現象で、遺伝子とその周辺の制御領域をまるごとコピーするので遺伝子の数が倍になります。脊椎動物の祖先で2回の全ゲノム重複が起こったと推測され、これにより、脊椎動物においては個々の遺伝子が別々に進化してさまざまな環境に対応して生き残れるようになり進化に有利に働いたと予想されています。
全ゲノムショットガン・アセンブリ
シークエンサー(DNA解読装置)が一回で読める長さは500~800塩基です。一方、ナメクジウオのゲノムサイズは約5億塩基、ヒトでは約30億塩基にも達するため、いったんゲノムDNAを断片化してからそれぞれの塩基配列を読みとり、それを編集してもとのゲノムDNAの構造を再構築する手法を全ゲノムショットガン法といいます。
- NHK(6月19日夜7時)で放送されました。
- 朝日新聞(6月19日 1面)、科学新聞(6月27日 4面)、京都新聞(6月19日 2面)、中日新聞(6月19日 28面)、日本経済新聞(6月19日 42面)、毎日新聞(6月19日 27面)および読売新聞(6月19日 33面)に掲載されました。