J. B. Brown 医学研究科講師らの研究チームは、複雑なAIやビッグデータを用いずに、高い精度で薬物の候補物質をスクリーニングする手法を開発しました。化合物の構造や過去の実験データから反応の予測に重要な組み合わせのみを選び、そのデータを用いて予測するもので、全実験データの10%から20%程度を使い、データベースに含まれる全ての化合物が、治療の標的となるタンパク質と反応するかどうかを高精度に予測することに成功しました。
本研究成果は、2017年3月6日午後9時に英国の学術誌「Future Medicinal Chemistry」に掲載されました。
研究者からのコメント

Brown講師
今回の研究は、製薬企業が持つ膨大なデータベースから精度良く新薬の候補となる化合物を発見する助けになると考えています。化合物に関する膨大なデータのごく一部から高精度な予測を立てることができたため、製薬企業ではどの化合物とタンパク質が予測精度を導くかを特定することが可能となりました。
この研究で用いたデータベースには少なくとも約4万点以上の化合物が含まれていました。今後、今回のモデルで数百万単位のデータを扱う際にも高精度で反応が予測できるのか検証し、実証研究も行い、創薬のコストダウンへ繋げていきたいと考えています。
概要
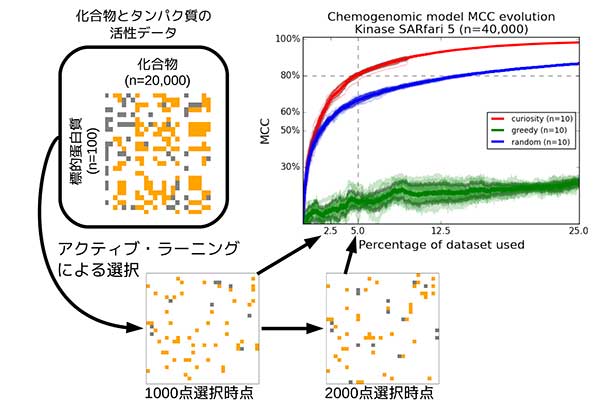
現在、世界各国で膨大な化合物のデータを用いた新薬の候補物質探索が行われています。数百万以上の化合物と疾患治療の標的となるタンパク質の反応を一つずつ調べるには膨大な資金と時間がかかるため、人工知能や数理モデルを用いて望ましい性質を持つ化合物を絞り込む必要があり、バイオインフォマティクスを用いた仮想スクリーニングへの注目が集まっています。DeepMind社のAlphaGoが囲碁のプロ棋士に勝利したこともあり、ディープラーニングや人工知能、ビッグデータ解析が注目を集めていますが、薬効の予測に関しては予測精度をわずかに上げるのに膨大なデータが必要だという課題があります。
そこで本研究グループは、いわゆるビッグデータ解析とは異なり、限られたデータから高精度の予測を実現する手法の開発を目指し、なるべくシンプルな予測モデルを構築しました。新薬開発の主な標的である細胞膜タンパク質の一種で、細胞内外の情報伝達を行うGタンパク質共役型受容体(GPCR)とキナーゼ(酵素)の計三つのデータベースを使いテストを行いました。
その結果、今回のモデルはどのデータベースでも高精度に、タンパク質と化合物が反応するかどうかを予測することができました。創薬全体のコスト削減やデータ解析の効率化への利用が期待されます。
詳しい研究内容について
書誌情報
【DOI】 http://doi.org/10.4155/fmc-2016-0197
【KURENAIアクセスURL】 http://hdl.handle.net/2433/218686
Daniel Reker, Petra Schneider, Gisbert Schneider & JB Brown. (2017). Active learning for computational chemogenomics. Future Medicinal Chemistry.
- 京都新聞(3月7日 29面)、日本経済新聞電子版(3月8日)に掲載されました。



